Parte 5: Filogenia Molecular
"No século 20, durante a década 70, os biólogos testaram as árvores filogenéticas, comparando as moléculas de várias espécies. Quanto mais semelhantes às moléculas de duas espécies diferentes, presumia-se que fossem mais aparentadas. Mas ao compararem mais e mais moléculas, descobriram - as moléculas diferentes davam resultados conflitantes. O padrão de árvore da vida baseada numa molécula contradizia o padrão obtido por outra. [James A. Lake, Ravi Jain e Maria C. Rivera, 'Mix and Match in the Tree of Life', Science 283 (1999), p. 2027-2028]. A árvore da vida, quem diria, está mais para 'gramado'..."
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
Aqui o autor do artigo erroneamente crê que o seu conhecimento adquirido sobre evolução molecular, é o que a teoria diz realmente.A questão começa pelo fato que, como a genética é estocástica (mutação, recombinação e reprodução sexuada são processos estocásticos e tem elemento probabilístico grande), a teoria da descendência comum não prediz que as árvores filogenéticas feitas com uma macromolécula se combinarão perfeitamente com qualquer outra árvore filogenética.Um exemplo óbvio disso é que uma criança pode ter olhos azuis, mesmo seu pai possuindo olhos marrons.O gene em um locus é diferente, mas nem por isso a criança não é seu descendente ou uma outra criança de olho marrom está mais relacionada com o pai do que com o seu filho.Além disso, em seqüências de DNA só existem quatros estados possíveis (A,T,G e C) e a chance de se encontrar um mesmo estado através de mutações aleatórias, em vez de um ancestral comum é de 25%.Em outros casos, temos a evolução convergente.Ruminantes não compartilham um ancestral comum recente com lângures apenas porque compartilham algumas mudanças de aminoácidos que não estão presentes em parentes próximos das duas espécies. A lisozima mudou a sua função durante a evolução em alguns herbívoros e agora tem função de digerir essas bactérias e liberar seus nutrientes.Incluir múltiplas macromoléculas para análise, está a solução para resolver este problema da análise da filogenia molecular (Avise & Wollenberg,1997).Por exemplo, um método utilizado é a hibridização DNA-DNA, onde a similaridade das seqüências de nucleotídeos é utilizada cadeias de DNA de duas espécies são separadas e depois reassociada formando duplex in vitro.Ela é o resultado estatístico das médias sobre bilhões de nucleotídeos e milhões de anos.A análise através do método utilizando as enzimas de restrição também é útil, assim como outros que não precisam ser mencionados aqui.
O que os biólogos moleculares utilizam como evidência para a teoria da descendência comum quando mostra, por exemplo, dados comparativos do Citocromo C de diferentes espécies são as evidências da redundância funcional protéica e do DNA que a codifica, não a comparação da árvore desenhada por uma macromolécula e a árvore filogenética morfológica mostrando congruência perfeita.Por exemplo, o gene que codifica o citocromo C do homem e do chimpanzé difere em míseros quatro nucleotídeos, sendo que existem 10 elevado a 49 seqüências diferentes que codificam essa proteína (Theobald,2004).E já se fez um estudo cuidadoso em que calculou que há um mínimo de 2.3 x 10 elevado a 93 seqüências funcionais possíveis da proteína do citocromo c, baseado em análises de mutações genéticas (Hampsey et al.,1988).Nos termos de uma análise estatística científica, "a hipótese nula" é que a identidade de aminoácidos não-essenciais nas proteínas do citocromo c do ser humano e do chimpanzé deve ser aleatória com respeito a um outro. Não há nenhuma razão a priori porque cada organismo deve ter as mesmas seqüências ou mesmo seqüências similares. Nenhuma seqüência específica é funcionalmente necessária em todo o organismo.Sem assumir a descendência comum, as seqüências de aminoácido e de proteínas seriam muito diferentes de cada um do outro.Se esse fosse caso, uma análise filogenética seria impossível, e esta forneceria a evidência muito forte para uma ancestralidade separada.
Outras evidências moleculares da ancestralidade comum podem ser citadas.Existem pseudogenes que são compartilhados entre espécies relacionadas filogeneticamente
,como o gene de globina ψη que compartilhamos com outros primatas na mesma posição do cromossomo, com as mesmas mutações que destroem sua função. Assim, uma vez que determinados organismos são encontrados que carregam o mesmo pseudogene, a teoria da descendência comum requer que todo os organismos filogeneticamente intermediários devem também carregar esse pseudogene. Suponha que nós descobríssemos que os seres humanos e os macacos de mundo velho compartilham de algum pseudogene redundante. De acordo com a descendência comum, todos os símios grandes (chimpanzés, gorilas, orangotangos, etc.) também deveriam necessariamente carregar o mesmo pseudogene redundante na mesma posição do cromossomo, uma vez que aparentemente não há mecanismos que removam pseudogenes em vertebrados.Além disso, compartilhamos instâncias de DNA viral nas mesmas posições genômicas em primatas com DNAs mais similares do que os nossos. Existem as instâncias do retrovírus endógeno que é conhecido como HERV-K e que são compartilhadas pela família dos primatas (Sverdlov, 2000).Seria incrível se alguns outros mamíferos (por exemplo: cães, vacas, etc.), tivessem estes mesmos retrovírus nas exatas mesmas posições cromossômicas.Se existisse uma vaca com os três retrogenes HERV-K que é original aos seres humanos, a teoria da descendência comum seria falsa, já que tais retrovírus não existem em outros primatas.Detalhe, chimpanzés possuem 11 DNAs do retrovírus HERV-K nas mesmas posições genômicas que nós ao passo que só compartilhamos 7 com gibões.Humanos e chimpanzés também compartilham em posições cromossômicas semelhantes “parasitas genômicos” chamados transpósons, que são sequências de DNAs que não são transcritas, mas podem mover para muitos sítios do genoma e ser re-inseridos.
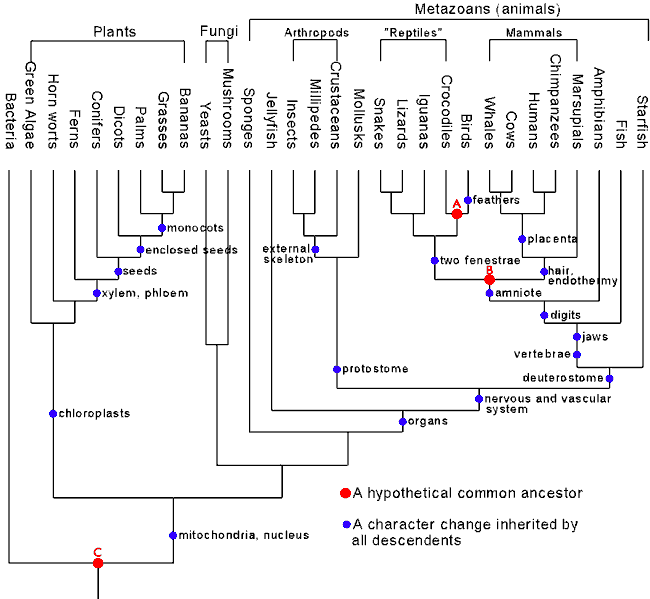
Sem dúvida, a evidência mais forte é a congruência entre dados filogenéticos independentes.Como é mais provável (partindo do pressuposto que a teoria da descendência comum está certa) que encontremos genes semelhantes em animais com morfologia semelhante do que um anatomicamente distinto, uma boa correlação entre as filogenias morfológicas e moleculares é um exemplo de teste envolvendo filogenias independentes.A "árvore filogenética padrão" neste gráfico do Talk Origins e que inclui 30 táxons apresenta filogenia molecular e morfológica convergindo de forma perfeita (Theobald,2004), mesmo havendo inacreditáveis 4,95 x 10 elevado a 38 árvores possíveis!!!Apesar disso, nem todas as árvores filogenéticas são perfeitamente congruentes.Mensuração de valores independentes em ciência nunca são exatas (como a constante física da carga do elétron ou a massa do próton).Quando mensurações independentes do mesmo valor entram em conflito, a questão relevante é perguntarmos: "Comparando-se duas mensurações, quanto de discrepância seria um problema para a teoria?".Essas questões são respondidas com estatísticas e probabilidades.O índice que mede a incongruência de filogenias independentes, o valor de P (que representa a razão do número máximo de árvores inconsistentes sobre o número total de árvores possíveis), fornece boa base para essas análises, assim como o valor de bootstrap, que estima a confiabilidade das árvores (ver parte 2 do artigo).Sendo assim, ainda que uma árvore dessas apresente incongruência, ela ainda pode ser capaz de apresentar congruência com elevado significado estatístico.A predição é que as árvores filogenéticas determinadas a partir das evidências moleculares e morfológicas devam se combinar com congruência significativa, caso contrário a teoria da descendência comum não faria sentido.Mas essa predição foi testada e corroborada inúmeras vezes na literatura científica (Penny et al, 1991).Também há evidência de congruência com significância estatística* entre as filogenias de grandes grupos eucariotos (Metazoa, Rodhophyta, Fungi,Euglenozoa,etc.) construídas por pequenas subunidades do RNA ribossômico e aquelas construídas conjuntamente por 4 proteínas (Baldauf et al,2000).Devemos considerar ainda que se a descendência comum fosse falsa, existiriam independentes filogenias com forte suporte apresentando nível de incongruência elevado (P>0,50).Como isso não ocorre (embora pudesse ocorrer), a teoria não só é falsificável como até agora não mostrou dados que a falseassem.
Todas filogenias , seja morfológica ou molecular, considera todos os amniotas são mais relacionado entre si que anfíbios. Em outro grupo, todos os répteis e aves são mais próximos entre si que são para mamíferos; finalmente, todas as aves e crocodilos são mais relacionados um com o outro que lagartos , cobras, e a tuatara (Gauthier, 1994). O único grupo que varia na posição por dados moleculares e morfológicos são as tartarugas.Esse é o fenômeno "árvore com longo ramo de atração" (Huelsenbeck & Hillis, 1993), que é quando um organismo teve a mudança evolucionária significativa que não pode facilmente ser comparado a outros organismos, e devido à natureza da metodologia usada avaliar a filogenia, ela pode parecer ser relacionado a muitos organismos possíveis.Árvores com longos ramos construídas pelo método da máxima parcimônia é um problema, porque quanto maior elas são, mais é provável que homoplasias se acumulem por mutação.Nestes casos, a exigência que se faz é que sejam utilizados genes com taxa de evolução vagarosa para minimizar este problema.De qualquer forma , um ou outro ramo inconsistente numa árvore não invalida a idéia da descendência comum.A inconsistência não é um problema, enquanto ela não for estatisticamente significante.
Algumas inconsistências alegadas simplesmente não existem, mas o Jonathan Wells acha um absurdo que a biologia molecular coloque vacas mais próximo de baleias do que de cavalos, mas ela é consistente de acordo com as evidências morfológicas e as confirmações através de descobertas de fósseis transicionais recentes,como as mostrada aqui na página da web de Thewissen . Artiodátilos (vacas, antílopes e porcos) não são mais relacionados a perissodátilos (cavalos, rinocerontes, etc) que baleias (Gingerich et al., 2001). Ele também fala que é um absurdo que equinodermos sejam um "grupo próximo" dos cordados.Isso revela desconhecimento sobre o trabalho de seus colegas de profissão.Taxonomistas englobam equinodermos e cordados como deuterostomados com clivagem radial e enterocefalia (Stearns & Hoekstra,2003.p.298).Além disso, em equinodermos, os genes ortólogos aos genes Hox de cordados são utilizados na padronização de estruturas com uma geometria diferente de qualquer outra encontrada nos demais filos, e mudanças nos fatores de transcrição organizou novas funções no desenvolvimento (Lowe & Wray,1997).Além disso, não se testa a teoria da descendência comum colocando apenas dois ou três táxons num cladograma.Sempre pode haver o caso de um ramo da árvore seja inconsistente, enquanto muitos outros comparados sejam consistentes e assim essa abordagem é errônea.
Enézio cita uma reportagem da Science em que os autores dizem que há tempos que se encontrou casos em que “o padrão da árvore da vida baseada numa molécula contradizia o padrão obtido por outra”, mas é dito lá que isto é um fato relevante para a teoria clônica da evolução do genoma eucarioto e não para a teoria da descendência comum.Esse artigo de Lake, Jain e Rivera pode ser lida num link da Bellarmin University.Teoria da descendência comum é uma coisa e a tese que diz os ancestrais dos primeiros eucariotos evoluíram quase que exclusivamente através de clones assexuados independentes (representada pela árvore da vida de Woesel) é outra.Para entendermos isso, passamos para outra hipótese abordada na reportagem, a hipótese dos três ramos principais (Eubactéria, Archae e Eucarya) que é baseada em uma filogenia construída por uma única molécula, seqüências de DNA codificadoras do RNA ribossômico.Recentemente, os genomas completos, de mais de uma dúzia de organismos foi descrito, e parte da história que eles nos contam criam algumas dificuldades a hipótese dos três ramos principais.Por exemplo, proteínas de eucariontes apresentam mais afinidades a Eubactéria que a Archae, aos quais se supõe que os Eucarya sejam mais relacionados.Algumas conclusões, tiradas de alguns genes, podem ser resolvidas postulando-se transferências gênicas horizontais entre as linhagens, um fenômeno relativamente comum em organismos unicelulares e que é observado regularmente nos dias atuais (Ochman et al,2000).Ainda que esses eventos de transferências gênicas fossem freqüentes o suficiente para sermos incapazes de apontar se existe três ramos principais ou uma rede mais complexa, isso não faz a menor diferença a teoria da descendência comum universal.Como em qualquer investigação científica, algumas condições devem ser cumpridas para que o resultado de uma observação seja confiável.Os métodos filogenéticos moleculares assumem que não há hibridização ou transferência por meio de vetores microbianos (Theobald,2004)- e isso não é, certamente, o caso de muitas bactérias.Inicialmente ,comparações baseadas em muitos locos sugeriam que ocorria pouca recombinação e que as populações de bactérias consistiam de clones independentes (Stearns & Hoekstra,2003,p.218).Posteriormente, o sequenciamento de DNA dos genes bacterianos revelou outra coisa: que cada cada gene pode ser constituído de porções diferentes de origens diferentes.Isso é uma consequência do "sexo localizado", ou seja, recombinação de trechos de DNA transferidos a células recipientes por plasmídios de conjugação, pela transformação ou transducção (Maynardi-Smith et al,1991).Consequentemente, se a premissa da transferência vertical de genes foi violada na evolução primitiva (coisa já observada nos unicelulares modernos), então a filogenia molecular não irá necessariamente recapitular a filogenia organísmica.É devido a isso que se diz que é questionável postular que a árvore construída por genes resolve a filogenia desses organismos (Doolittle,1999).
Além disso, se considerarmos o conceito biológico de espécie formulado por Ernst Mayr poderíamos dizer que as bactérias formam uma única "espécie global".Os protistas que reverteram formalmente o programa integrador de transferência gênica foram as primeiras espécies propriamente ditas, uma vez que a necessidade para se pertencer a uma espécie é a de se isolar reprodutivamente e ser potencialmente cruzante com outros indivíduos (Margulis & Sagan, 2002.p.146,147).Então, apontar o “último pool gênico comum” já é suficiente, uma vez que não há um grupo externo de controle para "enraizar" a árvore, como acontece quando enraizamos uma filogenia de tetrápodes colocando um peixe como grupo de controle. Assim, a árvore da vida não se origina de um único tronco, mas de um conjunto emaranhado de ramificações; nela, os organismos pioneiros em reverter o programa integrador de transferência gênica representam as primeiras bifurcações (Woese, 2000).
Entretanto, Jonathan Wells afirma a árvore da vida é uma conceito em crise porque a origem de todos os organismos a partir de um ancestral comum não pode ser postulada.Mas será que isso é importante?Vejamos o que Darwin diz na primeira edição de A Origem das Espécies:
"Therefore I cannot doubt that the theory of descent with modification embraces all the members of the same class. I believe that animals have descended from at most only four or five progenitors".(Darwin,1859)
Ou seja, Darwin considera que até mesmo no caso de todas as espécies da Terra serem descendentes de mais de uma forma primitiva, isso é uma situação que não contradiz sua teoria de descendência com modificação.É aí que temos que salientar que a árvore da vida darwiniana sequer supõe que um ancestral comum universal deva ser conhecido para que sua existência seja considerada válida.
*Nota bene:Este é o cladograma da filogenia dos eucariotos construídos conjuntamente por 4 proteínas.Está disponível neste link do site do Imperial College London .Já a filogenia contruída pelo método "multigene" é vista neste link do site da University and Jepson Herbaria of the University of California at Berkeley .Vejam que ,considerando as filogenias apresentadas, são 13 grupos eucariotos analisados por ambos os métodos.Foram 5 ramos inconsistentes encontrados.Assim, calculei que o valor de P ≤ 0,0095.Em termos estatísticos, isso significa uma congruência filogenética com alta significância estatística.
Referências:
Avise, J. C., and Wollenberg, K. (1997).Phylogenetics and the origin of species." PNAS 94: 7748-7755.Link: http://www.pnas.org/cgi/content/full/94/15/7748?ck=nck
Baldauf, S. L., Roger, A. J., Wenk-Siefert, I., and Doolittle, W. F. (2000).A kingdom-level phylogeny of eukaryotes based on combined protein data.Science 290: 972-7.
Darwin, C.(1859).The Origins of Species.Chapter 14: Recapitulation and Conclusion. Link:http://www.talkorigins.org/faqs/origin/chapter14.html
Doolittle, W. F. (1999).Phylogenetic Classification and the Universal Tree. Science 284: 2124. Link: http://cas.bellarmine.edu/tietjen/Ecology/phylogenetic_classification_and_.htm
Huelsenbeck, J. P. and D. M. Hillis (1993). Success of phylogenetic methods in the four-taxon case. Systematic Biology 42:247-264.
Gauthier, J. A.(1994). The diversification of the amniotes. In D. R. Prothro and R. M. Schoch eds. Major features of vertebrate evolution. Paleontological Society Short Courses in Paleontology 7:129-159.
Gingerich, P. D. et al. (2001). Origin of whales from early artiodactyls: Hands and feet of Eocene Protocetidae from Pakistan. Science 293: 2239-2242.
Hampsey, D. M., Das, G., and Sherman F. (1988).Yeast iso-1-cytochrome c: genetic analysis of structural requirements." FEBS Leters 231: 275-83.
Lowe C.J. and Wray,G.A.(1997).Radical alterations in the roles of homeobox genes during echinoderm evolution Nature 389:718-72.
Margulis, L. e Sagan, D. (2002).O Que é Vida?Jorge Zahar Editor, Rio de Janeiro-RJ.
Maynard-Smith, J., Dowson. C.G. and Spratt. B.G. (1991). Localized sex in bacteria. Nature 349: 29-31.
Ochman, H., Lawrence, J. G., Groisman, E. A. (2000) "Lateral gene transfer and the nature of bacterial innovation." Nature 405: 299-304.
Penny, D., Hendy, M. D. and Steel, M. A. (1991). Testing the theory of descent. In Phylogenetic Analysis of DNA Sequences, eds. Miyamoto, M. and Cracraft, J., Oxford University Press, New York. pp. 155-183.
Sverdlov, E. D. (2000).Retroviruses and primate evolution. BioEssays 22: 161-171.
Stearns S.C. e Hoekstra, R.F. (2003).Evolução: Uma Introdução.Atheneu Editora São Paulo, São Paulo-SP.
Theobald, Douglas L. “29+ Evidences for Macroevolution: The Scientific Case for Common Descent." The Talk.Origins Archive. Vers. 2.83. 2004. 12 Jan, 2004 <http://www.talkorigins.org/faqs/comdesc/>
Woese, C. R. (2000). Interpreting the universal phylogenetic tree. Proceedings of the National Academy of Science USA 97(15): 8392-8396.Link: http://www.pnas.org/cgi/content/abstract/97/15/8392
------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
Aqui o autor do artigo erroneamente crê que o seu conhecimento adquirido sobre evolução molecular, é o que a teoria diz realmente.A questão começa pelo fato que, como a genética é estocástica (mutação, recombinação e reprodução sexuada são processos estocásticos e tem elemento probabilístico grande), a teoria da descendência comum não prediz que as árvores filogenéticas feitas com uma macromolécula se combinarão perfeitamente com qualquer outra árvore filogenética.Um exemplo óbvio disso é que uma criança pode ter olhos azuis, mesmo seu pai possuindo olhos marrons.O gene em um locus é diferente, mas nem por isso a criança não é seu descendente ou uma outra criança de olho marrom está mais relacionada com o pai do que com o seu filho.Além disso, em seqüências de DNA só existem quatros estados possíveis (A,T,G e C) e a chance de se encontrar um mesmo estado através de mutações aleatórias, em vez de um ancestral comum é de 25%.Em outros casos, temos a evolução convergente.Ruminantes não compartilham um ancestral comum recente com lângures apenas porque compartilham algumas mudanças de aminoácidos que não estão presentes em parentes próximos das duas espécies. A lisozima mudou a sua função durante a evolução em alguns herbívoros e agora tem função de digerir essas bactérias e liberar seus nutrientes.Incluir múltiplas macromoléculas para análise, está a solução para resolver este problema da análise da filogenia molecular (Avise & Wollenberg,1997).Por exemplo, um método utilizado é a hibridização DNA-DNA, onde a similaridade das seqüências de nucleotídeos é utilizada cadeias de DNA de duas espécies são separadas e depois reassociada formando duplex in vitro.Ela é o resultado estatístico das médias sobre bilhões de nucleotídeos e milhões de anos.A análise através do método utilizando as enzimas de restrição também é útil, assim como outros que não precisam ser mencionados aqui.
O que os biólogos moleculares utilizam como evidência para a teoria da descendência comum quando mostra, por exemplo, dados comparativos do Citocromo C de diferentes espécies são as evidências da redundância funcional protéica e do DNA que a codifica, não a comparação da árvore desenhada por uma macromolécula e a árvore filogenética morfológica mostrando congruência perfeita.Por exemplo, o gene que codifica o citocromo C do homem e do chimpanzé difere em míseros quatro nucleotídeos, sendo que existem 10 elevado a 49 seqüências diferentes que codificam essa proteína (Theobald,2004).E já se fez um estudo cuidadoso em que calculou que há um mínimo de 2.3 x 10 elevado a 93 seqüências funcionais possíveis da proteína do citocromo c, baseado em análises de mutações genéticas (Hampsey et al.,1988).Nos termos de uma análise estatística científica, "a hipótese nula" é que a identidade de aminoácidos não-essenciais nas proteínas do citocromo c do ser humano e do chimpanzé deve ser aleatória com respeito a um outro. Não há nenhuma razão a priori porque cada organismo deve ter as mesmas seqüências ou mesmo seqüências similares. Nenhuma seqüência específica é funcionalmente necessária em todo o organismo.Sem assumir a descendência comum, as seqüências de aminoácido e de proteínas seriam muito diferentes de cada um do outro.Se esse fosse caso, uma análise filogenética seria impossível, e esta forneceria a evidência muito forte para uma ancestralidade separada.
Outras evidências moleculares da ancestralidade comum podem ser citadas.Existem pseudogenes que são compartilhados entre espécies relacionadas filogeneticamente
,como o gene de globina ψη que compartilhamos com outros primatas na mesma posição do cromossomo, com as mesmas mutações que destroem sua função. Assim, uma vez que determinados organismos são encontrados que carregam o mesmo pseudogene, a teoria da descendência comum requer que todo os organismos filogeneticamente intermediários devem também carregar esse pseudogene. Suponha que nós descobríssemos que os seres humanos e os macacos de mundo velho compartilham de algum pseudogene redundante. De acordo com a descendência comum, todos os símios grandes (chimpanzés, gorilas, orangotangos, etc.) também deveriam necessariamente carregar o mesmo pseudogene redundante na mesma posição do cromossomo, uma vez que aparentemente não há mecanismos que removam pseudogenes em vertebrados.Além disso, compartilhamos instâncias de DNA viral nas mesmas posições genômicas em primatas com DNAs mais similares do que os nossos. Existem as instâncias do retrovírus endógeno que é conhecido como HERV-K e que são compartilhadas pela família dos primatas (Sverdlov, 2000).Seria incrível se alguns outros mamíferos (por exemplo: cães, vacas, etc.), tivessem estes mesmos retrovírus nas exatas mesmas posições cromossômicas.Se existisse uma vaca com os três retrogenes HERV-K que é original aos seres humanos, a teoria da descendência comum seria falsa, já que tais retrovírus não existem em outros primatas.Detalhe, chimpanzés possuem 11 DNAs do retrovírus HERV-K nas mesmas posições genômicas que nós ao passo que só compartilhamos 7 com gibões.Humanos e chimpanzés também compartilham em posições cromossômicas semelhantes “parasitas genômicos” chamados transpósons, que são sequências de DNAs que não são transcritas, mas podem mover para muitos sítios do genoma e ser re-inseridos.
Sem dúvida, a evidência mais forte é a congruência entre dados filogenéticos independentes.Como é mais provável (partindo do pressuposto que a teoria da descendência comum está certa) que encontremos genes semelhantes em animais com morfologia semelhante do que um anatomicamente distinto, uma boa correlação entre as filogenias morfológicas e moleculares é um exemplo de teste envolvendo filogenias independentes.A "árvore filogenética padrão" neste gráfico do Talk Origins e que inclui 30 táxons apresenta filogenia molecular e morfológica convergindo de forma perfeita (Theobald,2004), mesmo havendo inacreditáveis 4,95 x 10 elevado a 38 árvores possíveis!!!Apesar disso, nem todas as árvores filogenéticas são perfeitamente congruentes.Mensuração de valores independentes em ciência nunca são exatas (como a constante física da carga do elétron ou a massa do próton).Quando mensurações independentes do mesmo valor entram em conflito, a questão relevante é perguntarmos: "Comparando-se duas mensurações, quanto de discrepância seria um problema para a teoria?".Essas questões são respondidas com estatísticas e probabilidades.O índice que mede a incongruência de filogenias independentes, o valor de P (que representa a razão do número máximo de árvores inconsistentes sobre o número total de árvores possíveis), fornece boa base para essas análises, assim como o valor de bootstrap, que estima a confiabilidade das árvores (ver parte 2 do artigo).Sendo assim, ainda que uma árvore dessas apresente incongruência, ela ainda pode ser capaz de apresentar congruência com elevado significado estatístico.A predição é que as árvores filogenéticas determinadas a partir das evidências moleculares e morfológicas devam se combinar com congruência significativa, caso contrário a teoria da descendência comum não faria sentido.Mas essa predição foi testada e corroborada inúmeras vezes na literatura científica (Penny et al, 1991).Também há evidência de congruência com significância estatística* entre as filogenias de grandes grupos eucariotos (Metazoa, Rodhophyta, Fungi,Euglenozoa,etc.) construídas por pequenas subunidades do RNA ribossômico e aquelas construídas conjuntamente por 4 proteínas (Baldauf et al,2000).Devemos considerar ainda que se a descendência comum fosse falsa, existiriam independentes filogenias com forte suporte apresentando nível de incongruência elevado (P>0,50).Como isso não ocorre (embora pudesse ocorrer), a teoria não só é falsificável como até agora não mostrou dados que a falseassem.
Todas filogenias , seja morfológica ou molecular, considera todos os amniotas são mais relacionado entre si que anfíbios. Em outro grupo, todos os répteis e aves são mais próximos entre si que são para mamíferos; finalmente, todas as aves e crocodilos são mais relacionados um com o outro que lagartos , cobras, e a tuatara (Gauthier, 1994). O único grupo que varia na posição por dados moleculares e morfológicos são as tartarugas.Esse é o fenômeno "árvore com longo ramo de atração" (Huelsenbeck & Hillis, 1993), que é quando um organismo teve a mudança evolucionária significativa que não pode facilmente ser comparado a outros organismos, e devido à natureza da metodologia usada avaliar a filogenia, ela pode parecer ser relacionado a muitos organismos possíveis.Árvores com longos ramos construídas pelo método da máxima parcimônia é um problema, porque quanto maior elas são, mais é provável que homoplasias se acumulem por mutação.Nestes casos, a exigência que se faz é que sejam utilizados genes com taxa de evolução vagarosa para minimizar este problema.De qualquer forma , um ou outro ramo inconsistente numa árvore não invalida a idéia da descendência comum.A inconsistência não é um problema, enquanto ela não for estatisticamente significante.
Algumas inconsistências alegadas simplesmente não existem, mas o Jonathan Wells acha um absurdo que a biologia molecular coloque vacas mais próximo de baleias do que de cavalos, mas ela é consistente de acordo com as evidências morfológicas e as confirmações através de descobertas de fósseis transicionais recentes,como as mostrada aqui na página da web de Thewissen . Artiodátilos (vacas, antílopes e porcos) não são mais relacionados a perissodátilos (cavalos, rinocerontes, etc) que baleias (Gingerich et al., 2001). Ele também fala que é um absurdo que equinodermos sejam um "grupo próximo" dos cordados.Isso revela desconhecimento sobre o trabalho de seus colegas de profissão.Taxonomistas englobam equinodermos e cordados como deuterostomados com clivagem radial e enterocefalia (Stearns & Hoekstra,2003.p.298).Além disso, em equinodermos, os genes ortólogos aos genes Hox de cordados são utilizados na padronização de estruturas com uma geometria diferente de qualquer outra encontrada nos demais filos, e mudanças nos fatores de transcrição organizou novas funções no desenvolvimento (Lowe & Wray,1997).Além disso, não se testa a teoria da descendência comum colocando apenas dois ou três táxons num cladograma.Sempre pode haver o caso de um ramo da árvore seja inconsistente, enquanto muitos outros comparados sejam consistentes e assim essa abordagem é errônea.
Enézio cita uma reportagem da Science em que os autores dizem que há tempos que se encontrou casos em que “o padrão da árvore da vida baseada numa molécula contradizia o padrão obtido por outra”, mas é dito lá que isto é um fato relevante para a teoria clônica da evolução do genoma eucarioto e não para a teoria da descendência comum.Esse artigo de Lake, Jain e Rivera pode ser lida num link da Bellarmin University.Teoria da descendência comum é uma coisa e a tese que diz os ancestrais dos primeiros eucariotos evoluíram quase que exclusivamente através de clones assexuados independentes (representada pela árvore da vida de Woesel) é outra.Para entendermos isso, passamos para outra hipótese abordada na reportagem, a hipótese dos três ramos principais (Eubactéria, Archae e Eucarya) que é baseada em uma filogenia construída por uma única molécula, seqüências de DNA codificadoras do RNA ribossômico.Recentemente, os genomas completos, de mais de uma dúzia de organismos foi descrito, e parte da história que eles nos contam criam algumas dificuldades a hipótese dos três ramos principais.Por exemplo, proteínas de eucariontes apresentam mais afinidades a Eubactéria que a Archae, aos quais se supõe que os Eucarya sejam mais relacionados.Algumas conclusões, tiradas de alguns genes, podem ser resolvidas postulando-se transferências gênicas horizontais entre as linhagens, um fenômeno relativamente comum em organismos unicelulares e que é observado regularmente nos dias atuais (Ochman et al,2000).Ainda que esses eventos de transferências gênicas fossem freqüentes o suficiente para sermos incapazes de apontar se existe três ramos principais ou uma rede mais complexa, isso não faz a menor diferença a teoria da descendência comum universal.Como em qualquer investigação científica, algumas condições devem ser cumpridas para que o resultado de uma observação seja confiável.Os métodos filogenéticos moleculares assumem que não há hibridização ou transferência por meio de vetores microbianos (Theobald,2004)- e isso não é, certamente, o caso de muitas bactérias.Inicialmente ,comparações baseadas em muitos locos sugeriam que ocorria pouca recombinação e que as populações de bactérias consistiam de clones independentes (Stearns & Hoekstra,2003,p.218).Posteriormente, o sequenciamento de DNA dos genes bacterianos revelou outra coisa: que cada cada gene pode ser constituído de porções diferentes de origens diferentes.Isso é uma consequência do "sexo localizado", ou seja, recombinação de trechos de DNA transferidos a células recipientes por plasmídios de conjugação, pela transformação ou transducção (Maynardi-Smith et al,1991).Consequentemente, se a premissa da transferência vertical de genes foi violada na evolução primitiva (coisa já observada nos unicelulares modernos), então a filogenia molecular não irá necessariamente recapitular a filogenia organísmica.É devido a isso que se diz que é questionável postular que a árvore construída por genes resolve a filogenia desses organismos (Doolittle,1999).
Além disso, se considerarmos o conceito biológico de espécie formulado por Ernst Mayr poderíamos dizer que as bactérias formam uma única "espécie global".Os protistas que reverteram formalmente o programa integrador de transferência gênica foram as primeiras espécies propriamente ditas, uma vez que a necessidade para se pertencer a uma espécie é a de se isolar reprodutivamente e ser potencialmente cruzante com outros indivíduos (Margulis & Sagan, 2002.p.146,147).Então, apontar o “último pool gênico comum” já é suficiente, uma vez que não há um grupo externo de controle para "enraizar" a árvore, como acontece quando enraizamos uma filogenia de tetrápodes colocando um peixe como grupo de controle. Assim, a árvore da vida não se origina de um único tronco, mas de um conjunto emaranhado de ramificações; nela, os organismos pioneiros em reverter o programa integrador de transferência gênica representam as primeiras bifurcações (Woese, 2000).
Entretanto, Jonathan Wells afirma a árvore da vida é uma conceito em crise porque a origem de todos os organismos a partir de um ancestral comum não pode ser postulada.Mas será que isso é importante?Vejamos o que Darwin diz na primeira edição de A Origem das Espécies:
"Therefore I cannot doubt that the theory of descent with modification embraces all the members of the same class. I believe that animals have descended from at most only four or five progenitors".(Darwin,1859)
Ou seja, Darwin considera que até mesmo no caso de todas as espécies da Terra serem descendentes de mais de uma forma primitiva, isso é uma situação que não contradiz sua teoria de descendência com modificação.É aí que temos que salientar que a árvore da vida darwiniana sequer supõe que um ancestral comum universal deva ser conhecido para que sua existência seja considerada válida.
*Nota bene:Este é o cladograma da filogenia dos eucariotos construídos conjuntamente por 4 proteínas.Está disponível neste link do site do Imperial College London .Já a filogenia contruída pelo método "multigene" é vista neste link do site da University and Jepson Herbaria of the University of California at Berkeley .Vejam que ,considerando as filogenias apresentadas, são 13 grupos eucariotos analisados por ambos os métodos.Foram 5 ramos inconsistentes encontrados.Assim, calculei que o valor de P ≤ 0,0095.Em termos estatísticos, isso significa uma congruência filogenética com alta significância estatística.
Referências:
Avise, J. C., and Wollenberg, K. (1997).Phylogenetics and the origin of species." PNAS 94: 7748-7755.Link: http://www.pnas.org/cgi/content/full/94/15/7748?ck=nck
Baldauf, S. L., Roger, A. J., Wenk-Siefert, I., and Doolittle, W. F. (2000).A kingdom-level phylogeny of eukaryotes based on combined protein data.Science 290: 972-7.
Darwin, C.(1859).The Origins of Species.Chapter 14: Recapitulation and Conclusion. Link:http://www.talkorigins.org/faqs/origin/chapter14.html
Doolittle, W. F. (1999).Phylogenetic Classification and the Universal Tree. Science 284: 2124. Link: http://cas.bellarmine.edu/tietjen/Ecology/phylogenetic_classification_and_.htm
Huelsenbeck, J. P. and D. M. Hillis (1993). Success of phylogenetic methods in the four-taxon case. Systematic Biology 42:247-264.
Gauthier, J. A.(1994). The diversification of the amniotes. In D. R. Prothro and R. M. Schoch eds. Major features of vertebrate evolution. Paleontological Society Short Courses in Paleontology 7:129-159.
Gingerich, P. D. et al. (2001). Origin of whales from early artiodactyls: Hands and feet of Eocene Protocetidae from Pakistan. Science 293: 2239-2242.
Hampsey, D. M., Das, G., and Sherman F. (1988).Yeast iso-1-cytochrome c: genetic analysis of structural requirements." FEBS Leters 231: 275-83.
Lowe C.J. and Wray,G.A.(1997).Radical alterations in the roles of homeobox genes during echinoderm evolution Nature 389:718-72.
Margulis, L. e Sagan, D. (2002).O Que é Vida?Jorge Zahar Editor, Rio de Janeiro-RJ.
Maynard-Smith, J., Dowson. C.G. and Spratt. B.G. (1991). Localized sex in bacteria. Nature 349: 29-31.
Ochman, H., Lawrence, J. G., Groisman, E. A. (2000) "Lateral gene transfer and the nature of bacterial innovation." Nature 405: 299-304.
Penny, D., Hendy, M. D. and Steel, M. A. (1991). Testing the theory of descent. In Phylogenetic Analysis of DNA Sequences, eds. Miyamoto, M. and Cracraft, J., Oxford University Press, New York. pp. 155-183.
Sverdlov, E. D. (2000).Retroviruses and primate evolution. BioEssays 22: 161-171.
Stearns S.C. e Hoekstra, R.F. (2003).Evolução: Uma Introdução.Atheneu Editora São Paulo, São Paulo-SP.
Theobald, Douglas L. “29+ Evidences for Macroevolution: The Scientific Case for Common Descent." The Talk.Origins Archive. Vers. 2.83. 2004. 12 Jan, 2004 <http://www.talkorigins.org/faqs/comdesc/>
Woese, C. R. (2000). Interpreting the universal phylogenetic tree. Proceedings of the National Academy of Science USA 97(15): 8392-8396.Link: http://www.pnas.org/cgi/content/abstract/97/15/8392
posted by espectro109 at 5:25 PM
![]()

{kind=link}
{kind=link}
2 Comments:
oakley sunglasses wholesale, christian louboutin outlet, replica watches, christian louboutin shoes, oakley sunglasses, uggs on sale, tiffany jewelry, burberry pas cher, michael kors pas cher, louis vuitton, chanel handbags, tiffany and co, longchamp outlet, cheap oakley sunglasses, ray ban sunglasses, christian louboutin uk, louis vuitton outlet, jordan pas cher, ray ban sunglasses, nike air max, nike air max, christian louboutin, louboutin pas cher, longchamp outlet, polo ralph lauren, prada handbags, ugg boots, oakley sunglasses, coach outlet, oakley sunglasses, nike free, longchamp pas cher, louis vuitton outlet, kate spade outlet, air max, tory burch outlet, jordan shoes, replica watches, ugg boots, sac longchamp pas cher, nike free run, nike outlet, kate spade, gucci handbags, ray ban sunglasses, prada outlet, longchamp outlet, polo outlet, polo ralph lauren outlet online
converse, p90x workout, herve leger, lululemon, hollister, north face outlet, instyler, karen millen uk, nfl jerseys, mont blanc pens, oakley, hermes belt, ghd hair, abercrombie and fitch, hollister, soccer jerseys, nike air max, new balance shoes, insanity workout, asics running shoes, vans outlet, chi flat iron, jimmy choo outlet, mac cosmetics, baseball bats, babyliss, ray ban, lancel, valentino shoes, montre pas cher, longchamp uk, soccer shoes, reebok outlet, beats by dre, hollister clothing, louboutin, ferragamo shoes, celine handbags, wedding dresses, timberland boots, bottega veneta, nike roshe run, nike air max, toms shoes, converse outlet, vans, ralph lauren, mcm handbags, north face outlet, gucci
Post a Comment
<< Home